Build and visualize annotated influenza phylogenies
To construct a phylogenetic tree, selected sequences in the analysis set and run "create phylogenetic tree". Building the tree can take up to a few minutes (don't attempt to build a tree from more than 200 sequences). The tree will be displayed in the browser, along with alignments of the nucleotide and amino acid sequences. When a segment has multiple ORFs/splice forms, the tree is based on the nucleotide sequence of that longest protein. In addition to the sequences selected by the user, the tree tool includes a number of related reference sequences to provide phylogenetic context.
The tree viewer
The screen shot below gives and overview over some of the features of nextflu tree viewer. The tree is annotated with amino acid changes that occur on different branches. The annotated tree can be downloaded as png or pdf images or as a newick tree file (buttons below the tree). For HA segments, the user can toggle (by ticking the box) between HA1/HA2 numbering according to the mature cleaved HA1/HA2 protein, or according to the full length HA0, starting with the initiating ATG codon.
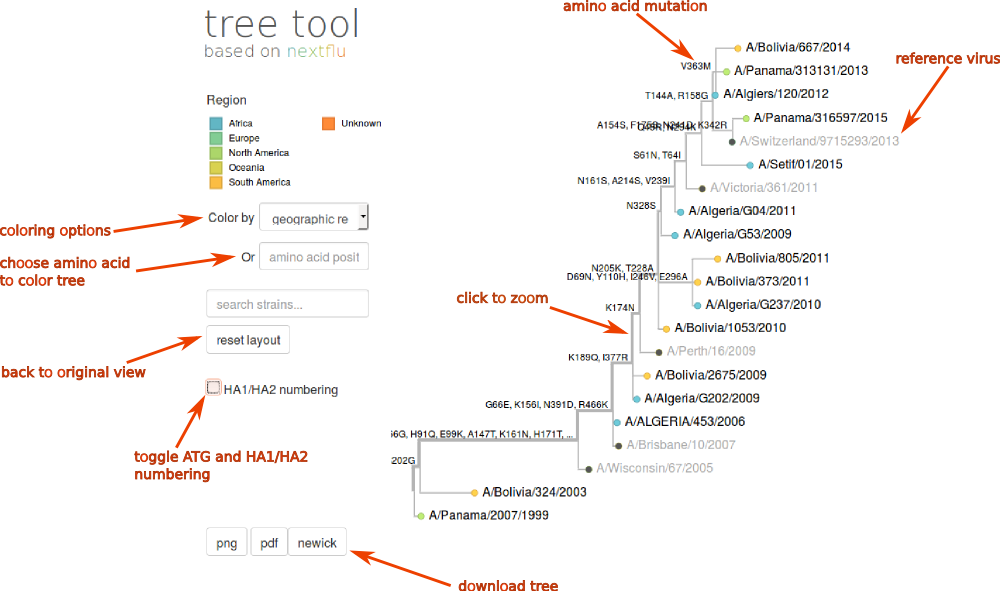
The tree can be coloured according to geographic region, sampling date, or virus subtype (as of now, passage and host group are not populated), using the pull down Color by menu, or according to amino acid identity at specific positions, by entering the position in the amino acid position box, or by clicking on the column number in the amino acid alignment below.
To zoom into clades on the tree, click on a branch; to zoom out, click reset layout. Labels of viruses corresponding to tips of the tree will show once the zoom level is high enough so that names are readable. Moving the mouse over a tip or a branch of the tree will show a small text box with additional information on the strain or the branch.
Results and download
The tree can can be downloaded as an image as PNG or PDF format or as newick tree file that can be displayed with other tree viewing and annotation programs. The amino acid mutations are included into the node labels. In some browsers, the newick file might be shown as text in the browser window. In this case, you should copy and paste the text into a text editor and save the file as a plain text file.
Alignment and tree building method
Sequences in the analysis sheet are aligned using MAFFT. The treetool than builds a phylogenetic tree using FastTree, which is then further refined using RAxML. Next, the state of every internal node of the tree is inferred using a marginal maximum likelihood method. Internal branches without mutations are collapsed into polytomies.
To root the tree, the tool searches a predefined list of reference sequences for the most similar sequence that predates the sequences to be analyzed by 3 years. If such a sequence is not available, the closest reference sequence is used. In addition to the sequences in the analysis sheet, a small number of reference sequences are added to the alignment to provide phylogenetic context.

