QIAGEN Influenza Annotation Pipeline in v2.0
Description
The annotation pipeline has been designed to run as a service via the CLC Genomics Workbench v8.5.1. A shell script has been provided that will, once configured, allow single- or batches of fasta sequences to be annotated in the manner described here.
The overall process is described schematically in the figures below each step below. There may be minor changes in the final pipeline compared to this diagram, and the pipeline is constantly undergoing testing and minor improvements as of the date of this document.
Briefly, the annotation pipeline has been designed to identify species, subtype, coding regions and various attributes from partial or complete consensus nucleotide sequences of varying quality derived from Influenza viruses in a robust and standardized manner. Only the following coding regions are identified:
- Influenza A: PB2,PB1,PB1-F2,PA,PA-X, HA,NP,NA,M1,M2,NS1,NS2
- Influenza B: PB2,PB1,PA,HA,NP,NA,NB,M1,BM2,NS1,NS2
- Influenza C: PB2,PB1,P3,HEF,NP,M1,CM2,NS1,NS2
Identify Segment Structure
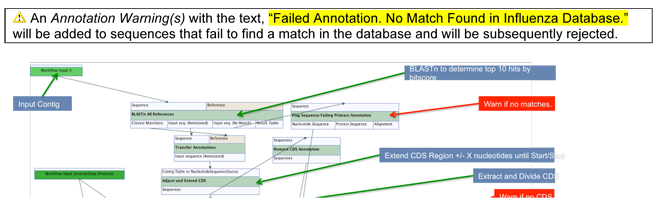
For Workflow Version 8.0.4.x and beyond, the entire query nucleotide sequence is matched first using BLAST1 to a curated and previously annotated nucleotide Influenza database where the top hit is identified by bitscore. A pairwise-alignment is subsequently made between the query and hit sequence and a sub-selection of annotations are transferred from the reference sequence to the query sequence. For example, the CDS and Mature peptide annotations are inherited directly from the match to the query sequence to define the segments overall gene structure.
The CDS region is subsequently adjusted at the terminal ends and will search for start and stop codons within a distance of X number of codons as determined by an adjustable parameter in the the pipeline itself.
Analyze Coding Regions
The adjusted CDS annotations from step 7.1 are extracted (maximum of two), sorted by length and by separated into two separate analysis paths. Should the CDS annotation be missing from the previous step, then the sequence will only report Segment/Subtype/Species annotations inherited in nucleotide space.
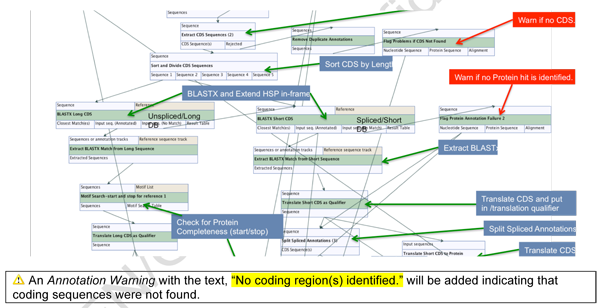
Should the annotation proceed, then each CDS path is analyzed using a modified BLASTx procedure to obtain the best hit from the protein database. The top BLASTx hit is extracted and partial HSP’s are extended to the length of the reference and annotated as BLASTX Match. The translation is placed into the BLASTX Match annotation and translation qualifier.
Start and stop codons are detected using patterns for each identified coding region on the the nucleotide query sequence. Only 100% matches are considered and start codons must be at the 5’ end of the sequence while stop codons must be at the 3’ end of the sequence to qualify
Identify Best Protein
The CDS sequences are divided into long and short and both are analyzed by BLASTx by translating each query nucleotide sequence in all six frames and matching to references obtained by re-curating and modifying components of the JCVI Vigor32 Influenza protein database. Due to the presence of short high-scoring pair (HSP) matches vs. reference databases, the reference results are elongated to full length for all BLASTx hits.
All CDS regions (and inherited annotations) are split IF they were originally spliced, then the split sequences are re-aligned to the query sequence and added as annotations.
The final step of the pipeline is the joining and extension of the spliced CDS regions and export of a .csv annotation file. No other annotations will be joined.
© QIAGEN 2013–17. All rights reserved

